Figure 1 from
Evidence for the widespread coupling of alternative splicing and nonsense-mediated
mRNA decay in humans.
Lewis BP, Green RE, Brenner SE,
Proc Natl Acad Sci U S A. 2003 Jan 7;100(1):189-92. [ pdf
]
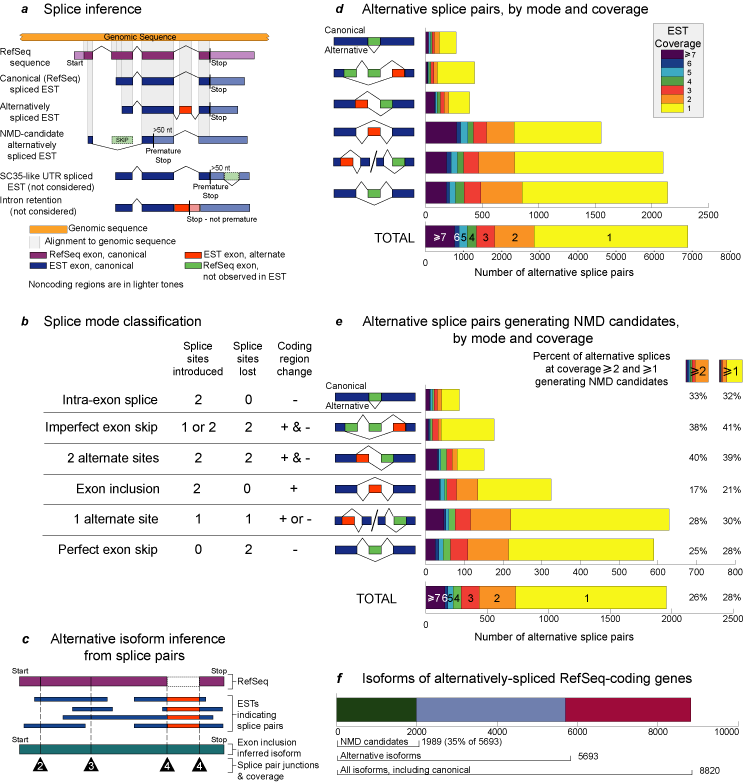
Alternative splice detection and classification. (a) Splice inference.
Coding regions of RefSeq mRNAs were aligned to the genomic sequence to
determine canonical splicing patterns. EST alignments to the genomic sequence
confirmed the canonical splices and indicated alternative splices. Canonical
(RefSeq) splices are indicated above the exons, whereas alternative splices
are indicated below the exons. When an alternative splice introduced a
stop codon >50 nucleotides upstream of the final exon-exon splice junction
of an inferred mRNA isoform, the stopcodon was classified as a premature
termination codon and the corresponding mRNA isoform was labeled a NMD
candidate. In the NMD-candidate example shown, an exon skip caused a frameshift,
resulting in the introduction of a premature termination codon. Restricting
the analysis to coding regions assured high alignment quality, but this
excluded alternative splicing in noncoding regions, such as that which
occurs with splicing factor SC35. Intron retentions were also excluded
because ESTs indicating intron retention are indistinguishable from incompletely
processed transcripts, a common dbEST contaminant. (b) Splice mode
classification. Alternative splices were categorized according to splice
site usage and effects on the coding sequence. "Splice sites introduced"
shows the number of splice donor/acceptor sites that were observed in the
alternative splice but were not included in the canonical splice. "Splice
sites lost" shows the number of splice donor/acceptor sites that were included
in the canonical splice but were absent in the alternative splice. "Coding
region change" indicates whether an alternative splice added (red) or subtracted
(green) coding sequence to the alternative isoform relative to the canonical
isoform. By our method, mutually exclusive exon usage appears as exon inclusion.
Our analysis excluded intron retentions, which would be classified as zero
splice sites introduced, two sites lost, and addition of coding sequence.
(c) Alternative isoform inference from splice pairs. Splice pairs
are splice donor/acceptor sites (![]() )
inferred from the alignments. Alternative splice pairs are those indicated
by ESTs, but not by a RefSeq mRNA. The exon composition of an isoform was
determined from EST-demonstrated splice pairs, which may be covered by
multiple ESTs. Coverage of splice pairs is indicated in each
)
inferred from the alignments. Alternative splice pairs are those indicated
by ESTs, but not by a RefSeq mRNA. The exon composition of an isoform was
determined from EST-demonstrated splice pairs, which may be covered by
multiple ESTs. Coverage of splice pairs is indicated in each ![]() .
Coverage for a complete isoform is not meaningful because of the variability
in coverage of its splice pairs. (d) Alternative splice pairs by
mode and coverage. The total number of alternative splice pairs associated
with each splicing mode is shown at various levels of EST coverage. The
distance from the
y axis to the right edge of each box corresponds
to the total number of splice pairs with coverage greater than or equal
to the number indicated. Note that each exon inclusion event involves two
splice pairs. (e) Alternative splice pairs generating NMD candidates
by mode and coverage. The panel shows the subset of alternative splice
pairs that produce premature termination codons. These splice pairs are
involved in generating NMD-candidate mRNA isoforms. The numbers of splice
pairs are displayed as in d. Also shown are the NMD-candidate splice
pairs at coverage >=1 and >=2 as a percentage of all alternative splice
pairs for each splicing mode. (f)Isoforms of alternatively spliced
RefSeq-coding genes. Shown are the total numbers of isoforms of theRefSeq-coding
genes for which alternative isoforms were found. These are subdivided into
the following categories: all isoforms including canonical, alternative
isoforms (i.e., all isoforms excluding canonical), and NMD candidates.
.
Coverage for a complete isoform is not meaningful because of the variability
in coverage of its splice pairs. (d) Alternative splice pairs by
mode and coverage. The total number of alternative splice pairs associated
with each splicing mode is shown at various levels of EST coverage. The
distance from the
y axis to the right edge of each box corresponds
to the total number of splice pairs with coverage greater than or equal
to the number indicated. Note that each exon inclusion event involves two
splice pairs. (e) Alternative splice pairs generating NMD candidates
by mode and coverage. The panel shows the subset of alternative splice
pairs that produce premature termination codons. These splice pairs are
involved in generating NMD-candidate mRNA isoforms. The numbers of splice
pairs are displayed as in d. Also shown are the NMD-candidate splice
pairs at coverage >=1 and >=2 as a percentage of all alternative splice
pairs for each splicing mode. (f)Isoforms of alternatively spliced
RefSeq-coding genes. Shown are the total numbers of isoforms of theRefSeq-coding
genes for which alternative isoforms were found. These are subdivided into
the following categories: all isoforms including canonical, alternative
isoforms (i.e., all isoforms excluding canonical), and NMD candidates.