About StrVCTVRE
StrVCTVRE predicts the pathogenicity of deletions and duplications that overlap an exon in the human genome. Just as there are methods to predict the pathogenicity of missense variants (such as REVEL, CADD, and many others), StrVCTVRE predicts the pathogenicity of exon-affecting deletions and duplications. StrVCTVRE was developed by researchers at UC Berkeley and Harvard Medical School. On this website, you can score a single variant with StrVCTVRE, annotate a VCF or BED file with StrVCTVRE, or download StrVCTVRE to run offline.

How StrVCTVRE works
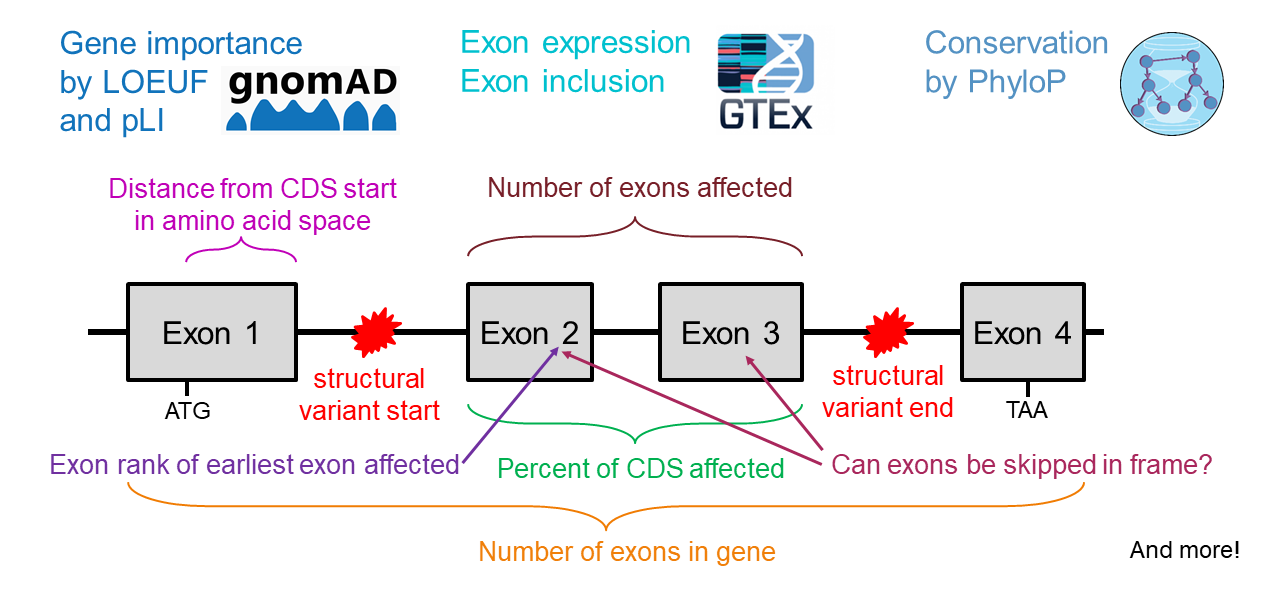
StrVCTVRE learns to identify pathogenic variants using a random forest trained on data from ClinVar, gnomAD, and a recent great ape sequencing study. StrVCTVRE integrates features that capture gene importance, coding regions, conservation, expression, and exon structure. StrVCTVRE performs accurately across a wide SV size range on independent test sets. We anticipate clinicians and researchers will use StrVCTVRE to prioritize structural variants identified during exome or genome sequencing in rare disease probands where no structural variant is immediately compelling.