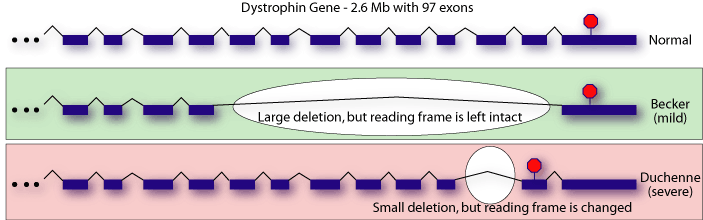
Once it was discovered that Duchenne MD and Becker
MD were both forms of the same disease, researchers attempted to determine
which regions of the dystrophin gene were most important by correlating
genotype and phenotype. It was mystifying that several large deletions
were present in patients with the mild, Becker, form, while smaller deletions
were sometimes found in patients with Duchenne MD[1-4].
What seemed to be more important than the amount of coding sequence that was deleted was whether or not the offending mutation resulted in a frameshift or not. Researchers working on the mouse model of Duchenne MD, mdx mouse, found a similarly puzzling result: a C-terminally truncated version of dystrophin was competent to rescue the MD phenotype in these mice[5]. In 1988, it was entirely unclear how to explain the apparent disconnect between genotype and phenotype.
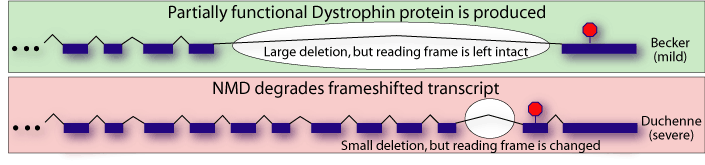
In light of NMD, these findings are comprehensible. Becker MD patients have a partially functional version of dystrophin that leads to less severe symptoms. Duchenne MD patients, however, have mutations that prevent any functional dystrophin production. Because of NMD, this includes mutations that introduce premature termination codons.
Appreciation of the role of NMD in MD has led to improved diagnostics and promising new treatment strategies. For example, the drug gentamicin, which causes translational read-through of stop codons, has shown some effectiveness for restoring dystrophin expression in cultured cells from mdx-mice[6]. Human clinical trials are currently underway.
[1]E. F. Gillard, J. S. Chamberlain, E. G. Murphy, C. L. Duff, B. Smith, A. H. Burghes, M. W. Thompson, J. Sutherland, I. Oss, S. E. Bodrug, and et al., "Molecular and phenotypic analysis of patients with deletions within the deletion-rich region of the Duchenne muscular dystrophy (DMD) gene," Am J Hum Genet, vol. 45, pp. 507-20, 1989.
[2]M. Koenig, A. H. Beggs, M. Moyer, S. Scherpf, K. Heindrich, T. Bettecken, G. Meng, C. R. Muller, M. Lindlof, H. Kaariainen, and et al., "The molecular basis for Duchenne versus Becker muscular dystrophy: correlation of severity with type of deletion," Am J Hum Genet, vol. 45, pp. 498-506, 1989.
[3]R. G. Roberts, R. J. Gardner, and M. Bobrow, "Searching for the 1 in 2,400,000: a review of dystrophin gene point mutations," Hum Mutat, vol. 4, pp. 1-11, 1994.
[4]T. P. Kerr, C. A. Sewry, S. A. Robb, and R. G. Roberts, "Long mutant dystrophins and variable phenotypes: evasion of nonsense-mediated decay?," Hum Genet, vol. 109, pp. 402-7, 2001.
[5]G.E. Crawford, J.A. Faulkner, R.H. Crosbie, K.P. Campbell, S.C. Froehner, and J.S. Chamberlain, "Assembly of the dystrophin-associated protein complex does not require the dystrophin COOH-terminal domain," J Cell Biol, vol. 150, pp. 1399-410, 2000.
[6]E. R. Barton-Davis, L. Cordier, D. I. Shoturma, S. E. Leland, and H. L. Sweeney, "Aminoglycoside antibiotics restore dystrophin function to skeletal muscles of mdx mice," J Clin Invest, vol. 104, pp. 375-81, 1999.
For more information about Muscular Dystrophy:
Muscular Dystrophy Association USA
Duchenne Muscular
Dystrophy:Research Approaches Toward a Cure (from Testlaboratorium
Breitnau)